
总部咨询热线
132-808-33289

专业服务

精益求精

专业讲师

良好口碑

西美咨询15年专注验厂、认证服务。已在青岛、上海、苏州、杭州、宁波、武汉、深圳等地设立30+分公司及分支机构,全国就近安排,本地化服务。
自公司成立以来,一直立足于客户实际,注重服务效果,全心全意为客户服务,至今已成功服务各类企业近万家,帮助企业优化流程、提高质量、破除贸易壁垒、赢得全球市场!
全国咨询热线
132-808-33289▉ MDR适用产品范围
1)常规医用器械(I/IIa/IIb/III 类):口罩、敷料、注射器、手术器械、监护仪、呼吸机、骨科植入物、心脏瓣膜、牙科器械、医用软件 SaMD、康复设备等;
2)有源植入器械:心脏起搏器、神经刺激器、植入式给药装置等;
3)Annex XVI 无医疗宣称类医美器械(强制纳入 MDR 管控):塑形假体、美瞳隐形眼镜、减脂仪器、皮肤填充剂、医美射频设备等;
4)配套耗材:器械清洁、消毒、灭菌产品、医疗器械配件、包装组件。
")
▉ MDR覆盖市场主体
境外医疗器械制造商、品牌方、研发企业;
欧盟境内进口商、分销商、仓储服务商;
零部件供应商、委托代工厂、临床试验机构。
豁免品类:体外诊断器械(执行 IVDR)、药品、活细胞 / 组织类产品、军工国防专用设备。
▉ MDR器械风险等级
依据 MDR 附录 VIII 分类规则,按使用风险划分为 4 个等级,审核难度逐级提升:
Class I 低风险:普通纱布、一次性镊子、基础护理器械;可自我宣告,无需公告机构 NB 介入;
Class IIa 中低风险:牙科充填材料、雾化器、无创监护设备;需公告机构审核体系 + 技术文件;
Class IIb 中高风险:呼吸机、激光治疗设备、植入式骨科耗材;全流程严格审核,含临床评价深度核查;
Class III 高风险:心脏支架、人工瓣膜、神经植入器械、长效医美填充产品;公告机构全文档审查 + 现场双阶段审核,临床证据要求极高。
▉ MDR八大核心强制合规要求
(一)ISO 13485 质量管理体系落地
企业必须建立符合 ISO 13485:2016 完整医疗器械质量体系,覆盖设计开发、采购、生产、检验、不良事件、变更控制;公告机构分一阶段文件审核、二阶段现场全流程审核,体系为取证基础门槛。
(二)全套完整技术文档 TEF(附录 II/III 强制)
技术档案是 MDR 审核核心,文件体量远高于旧 MDD,必备模块:
1. 器械分类判定报告、风险管理全流程文件;
2. 设计开发历史文件 DHF、验证 / 确认测试报告;
3. 标签、说明书、包装合规资料;
4. 原材料、零部件供应商管控记录;
5. 临床评价报告 CER、上市后监控 PMS 计划;
6. 生物相容性、化学有害物质、电气安全等检测报告。
(三)临床证据强制升级(MDR 最大难点)
1. 禁止单纯沿用文献,需匹配产品真实临床数据;高风险 III/IIb 类器械需充分临床试验或等同器械充分临床比对;
2. 建立上市后监控 PMS 机制,定期出具PSUR 定期安全更新报告:III 类器械每 6 个月提交,低风险器械周期最长不超过 2 年;
3. 建立不良事件警戒系统,出现严重风险需 24 小时内上报欧盟主管机构。
(四)UDI 唯一器械标识数字化溯源
全品类分阶段强制执行 UDI 二维码标识,录入欧盟 Eudamed 数据库:
- III 类器械:2021.5.26 起强制;
- IIa/IIb 类器械:2023.5.26 起强制;
- I 类器械:2025.5.26 起强制;
每件器械唯一编码,实现生产、流通、召回全链条一键溯源。
")
(五)Eudamed 欧盟医疗器械数据库全注册
企业、器械、欧盟授权代表、UDI、不良事件、证书信息全部线上录入 Eudamed,公开可查;未完成注册产品禁止投放欧盟市场。
(六)欧盟授权代表 EU REP 硬性要求
所有境外厂商必须指定欧盟境内授权代表,全权对接监管机构、接收投诉、保管全套技术文档,承担连带责任,无 EU REP 无法清关销售。
(七)上市后持续监管与召回机制
1. 建立产品全生命周期追溯台账,可快速完成全批次召回;
2. 公告机构每年监督审核,每 5 年完整复审换证;
3. 欧盟主管机构可开展无预警飞行检查,核查现场、文件、临床记录。
(八)有害物质与生物安全管控
严控邻苯、重金属、致癌助剂、纳米材料风险;植入类器械强制全套生物相容性测试,符合 ISO 10993 系列标准。
▉ MDR关键时间节点
")
2017.05.05:MDR 法规正式发布;
2021.05.26:MDR 全面强制实施;
2025.05.26:I 类器械 UDI 标识强制落地;
过渡期政策全部关闭,原 MDD 旧证书已全部失效,所有在售器械必须完成 MDR 转换。
▉ MDR违规处罚后果
1. 货物欧盟海关直接扣留、整批销毁 / 退回,产生高额物流、滞港损失;
2. 产品全欧盟市场下架、强制召回;
3. 高额罚款,最高可达企业全球年营业额 4%;
4. 吊销 CE 证书,永久限制企业欧盟市场准入;
5. 引发医疗纠纷、品牌口碑崩塌,丢失海外长期客户订单。
▉ MDR服务流程
西美咨询|一站式 MDR 合规全流程服务
针对医械、医美企业 MDR 合规难点,提供全程落地辅导,特色服务:
1. 前期差距诊断
判定器械风险等级,逐条对标 MDR 条款梳理短板,出具定制化整改时间表。
2. 体系全套搭建
辅导落地 ISO13485 体系,编制手册、程序文件、SOP、全套记录表单。
3. 技术档案专项编制
专业团队撰写风险管理报告、CEP 临床评价计划、CER 临床报告、GSPR 符合性论证、标签说明书全套资料。
4. UDI&Eudamed 注册协助
指导 UDI 编码设计、标签改版,代办企业 SRN 注册号、器械信息数据库录入。
5. 公告机构对接 + 全程陪审
开通机构绿色通道,缩短排期;审核现场专人全程陪同,实时答疑,快速闭环不符合项,提升一次性通过率。
6. 长效维护服务
上市后 PMS/PMCF 体系搭建、年度监督审核辅导、法规更新同步,持续保障证书有效。
如需了解MDR欧盟医疗器械法规服务流程及费用,欢迎联系西美技术服务。
服务热线:132-808-33289 | 官网:https://www.ximeicsr.com









RSCI(Responsible Supply Chain Initiative)于2021年10月由汽车行业的多家公司和协会共同成立,旨在共同制定统一的评估标准,支持汽车行业及相关领域的成员、供应商和利益相关者构建更加负责任的供应链体系。为实现这一目标,RSCI向成员提供工具、措施与一般支match持,以推动行业履行社会责任与尽职调查义务。
欧盟史上最严包装法规 PPWR(EU 2025/40)将于2026 年 8 月 12 日全面强制实施,彻底替代旧包装指令,所有出口欧盟、跨境电商卖家必须完成各国 EPR 生产者责任延伸注册。无合规注册号、未按期申报缴费将面临链接下架、海关扣货、最高年营业额 6% 高额处罚。
全球领先的环境信息披露平台 CDP 发布 2026 年披露季问卷重要变更,本次调整围绕自然披露拓展、标准框架对齐、中小企业支持等核心方向展开,新增多个披露主题、优化评分体系并简化披露流程,旨在为资本市场和供应链提供更全面、可比的环境数据。无论是参与 CDP 披露的大型企业还是中小企业,都需要重点关注这些变化,提前做好准备。
2023年8月,新的《欧盟电池与废电池法规》正式生效,于2024年2月18日起实施。该法规适用于在欧盟本地生产及进口的电池。本期针对大家关心的欧盟电池法规标签最新要求为大家答疑解惑。希望通过本次解答,助力各企业精准把握法规要求,找到优质高效的应对路径,更从容地应对合规挑战。

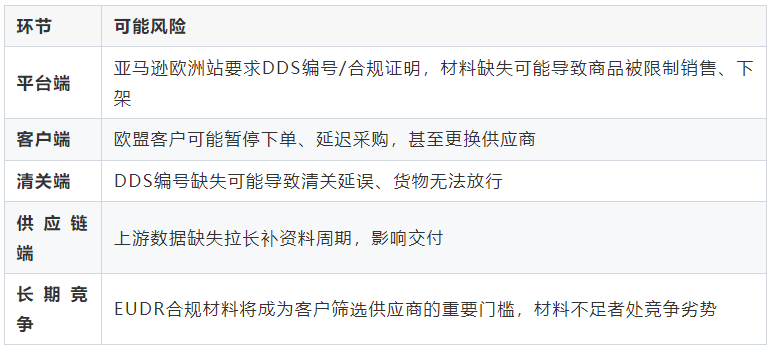



































")
")
")
